This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison
Conclusions
Lynch Syndrome and MSH6
Lynch syndrome is also know as hereditary nonpolyposis colorectal cancer and is a hereditary cancer disorder that leads to increased risk of developing many types of cancer, but primarily colorectal and endometrial cancers [1]. Lynch syndrome is caused by a mutation in a DNA mismatch repair gene which leads to an accumulation of mutations which contributes to the development of cancer [2]. MSH6 is a gene involved in the DNA mismatch repair where it recognizes mismatched bases and recruits other factors to replace the incorrect base [3].
MSH6 has three domains; a PWWP domain that helps it associate with PCNA and DNA, and two MUTS domains that help it do its mismatch repair function. MSH6 is involved in the mismatch repair pathway and has DNA binding function. It is located in the nucleus as that is where DNA is located. MSH6 is well conserved between species, especially in the MUTS domains. The PWWP domain is only conserved in mammals.
MSH6 has three domains; a PWWP domain that helps it associate with PCNA and DNA, and two MUTS domains that help it do its mismatch repair function. MSH6 is involved in the mismatch repair pathway and has DNA binding function. It is located in the nucleus as that is where DNA is located. MSH6 is well conserved between species, especially in the MUTS domains. The PWWP domain is only conserved in mammals.
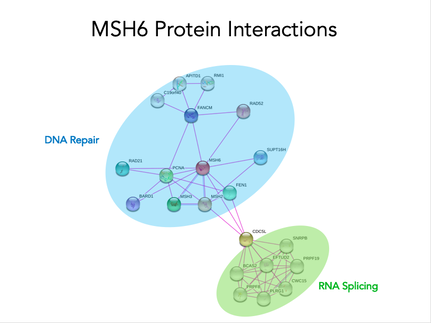
MSH6 interacts with many proteins involved in DNA repair and not just proteins in the mismatch repair pathway, but other DNA repair pathways as well. Additionally, it interacts with proteins involved in RNA binding and splicing.
Estrogen signaling stimulates cell proliferation in the endometrium. This occurs naturally as part of the menstrual cycle, but the more exposure to estrogen one has the greater the risk of developing endometrial cancer [4]. Increased estrogen exposure can result from having more body fat (as adipose tissue secretes estrogen), hormone therapies for menopause or breast cancer, as well as longer exposure to estrogen from early menstruation, late menopause, or never being pregnant. [5]
Mice make a good model organism for studying endometrial cancer as they have a similar reproductive system to humans. Mice with mutated MSH6 have been shown to develop endometrial cancer [6].
Despite all of this knowledge, it is unexplored how estrogen receptor signaling in cells with MSH6 mutation contributes to the development of endometrial cancer. My primary goal is to determine whether and how estrogen receptor alpha (ERα) signaling in cells with an MSH6 mutation contributes to the development of endometrial cancer. My hypothesis is that MSH6 is involved in the activation of ERα as a transcription factor leading to endometrial cell proliferation.
Despite all of this knowledge, it is unexplored how estrogen receptor signaling in cells with MSH6 mutation contributes to the development of endometrial cancer. My primary goal is to determine whether and how estrogen receptor alpha (ERα) signaling in cells with an MSH6 mutation contributes to the development of endometrial cancer. My hypothesis is that MSH6 is involved in the activation of ERα as a transcription factor leading to endometrial cell proliferation.
Aim 1: Identify ERα interaction motifs in MSH6
In order to find locations at which estrogen receptors interact with MSH6, I will align MSH6 sequences between many species. I will compare the sequences of species with estrogen and species without estrogen. I predict that sites of ERα interaction will be conserved in species that have estrogen, but not conserved in species without estrogen.

I will also use bioinformatic tools such as MEME to predict possible ERα interaction motifs within MSH6. I will then test whether these predicted locations bind ERα by mutating them and performing an immunoprecipitation assay. To determine how ERα interaction contributes to development of endometrial cancer, I will mutate the sites of interaction in mice using CRISPR/Cas9 and then see if the animals develop endometrial cancer.

Aim 2: Determine how estrogen interaction with MSH6 affects the transcriptome
Estrogen receptors act as transcription factors to regulate gene expression in response to estrogen [7]. To determine how ERα interaction with MSH6 affects transcription of genes, I will compare wild-type mice and mice with MSH6 knockout with and without estrogen. To control the amount of estrogen mice receive, I will perform an oophorectomy on the mice, as the ovary is the primary estrogen producing organ, and without it levels of estrogen are significantly reduced. I will perform RNA-seq on endometrial tissue of these four groups of mice and then use gene ontology to determine what biological processes are affected by exposure to estrogen. I predict that genes involved in cell proliferation and evasion of apoptosis will be up-regulated in response to estrogen, and this will become dysregulated with MSH6 mutation.
Aim 3: Determine how estrogen interaction with MSH6 impacts protein expression
MSH6 interacts with many proteins involved in RNA binding and splicing so it could also be regulating gene expression post-transcriptionally. In order to test this hypothesis, I will take the wild-type and MSH6 knockout mice and expose some to estrogen and some to no estrogen and compare these to a wild type SILAC mouse. I will compare the protein levels between these groups and use gene ontology to determine which biological processes are being affected. I predict that proteins involved in cell proliferation and evasion of apoptosis will have higher levels with exposure to estrogen and this will become dysregulated with MSH6 mutation.
Future Directions
Currently the main way to prevent endometrial cancer in individuals at a high risk of developing it, such as individuals with Lynch syndrome, is to perform a hysterectomy [5]. Individuals with Lynch syndrome are more likely to develop cancer at a younger age than the general population, this means that individuals may still be in their child-bearing years when they are at risk for developing endometrial cancer [8]. Understanding the specific mechanism by which estrogen is affecting the development of endometrial cancer in individuals with Lynch syndrome can help determine potential hormone therapies that can be used in its prevention and treatment. Estrogen is protective against colorectal cancer so hormone therapy used to reduce the risk of endometrial cancer would have to be done very precisely in order to avoid losing the protective effects estrogen provides [9].
References:
[1] Lynch, H (2005). What the Physician Needs to Know About Lynch Syndrome:An Update. Oncology 19 (4) 455-463. Retrieved from http://www.cancernetwork.com/colorectal-cancer/what-physician-needs-know-about-lynch-syndrome-update/page/0/1
[2] Lynch, H, Smryk, T, & Lynch, J. (1997) An update of HNPCC (Lynch Syndrome). Cancer Genetics and Cytogenetics 9(1): 84-99. doi: 10.1016/S0165-4608(96)00290-7
[3] Marsischky, GT, Filosi, N, Kane, MF, Kolodner, R. (1996) Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes & Development 10(4): 407-420. doi: 10.1101/gad.10.4.407
[4] Ziel, HK. (1982). Estrogen’s role in endometrial cancer. Obstet Gynecol. 60(4): 509-15. Retrieved from: https://ovidsp-tx-ovid-com.ezproxy.library.wisc.edu/sp-3.33.0b/ovidweb.cgi?WebLinkFrameset=1&S=MMLEFPOFPEDDEENBNCDKGBJCNEPDAA00&returnUrl=ovidweb.cgi%3fMain%2bSearch%2bPage%3d1%26S%3dMMLEFPOFPEDDEENBNCDKGBJCNEPDAA00&directlink=https%3a%2f%2fovidsp.tx.ovid.com%2fovftpdfs%2fFPDDNCJCGBNBPE00%2ffs047%2fovft%2flive%2fgv038%2f00006250%2f00006250-198210000-00022.pdf&filename=ESTROGEN%27S+ROLE+IN+ENDOMETRIAL+CANCER.&navigation_links=NavLinks.S.sh.22.1&link_from=S.sh.22%7c1&pdf_key=FPDDNCJCGBNBPE00&pdf_index=/fs047/ovft/live/gv038/00006250/00006250-198210000-00022&D=ovft&link_set=S.sh.22|1|sl_10|resultSet|S.sh.22.23|0
[5] PDQ Screening and Prevention Editorial Board. (Updated 2019, March 15). PDQ Endometrial Cancer Prevention. National Cancer Institute. Retrieved from: https://www.cancer.gov/types/uterine/patient/endometrial-prevention-pdq Accessed 05/01/2019
[6]de Wind, N., Dekker, M., Claij, N., Jansen, L., van Klink, Y., Radman, M., Riggins, G., van der Valk, M., van’t Wout, K., & te Riele, H. (1999). HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nature Genetics 23: 359-362 doi: 10.1038/15544
[7] Frasor, J., Danes, J., Komm, B., Chang, K., Lyttle, C., & Katzenellenbogen, B. (2003) Profiling of estrogen up- and down- regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 144(10): 4562-4574. doi: 10.1210/en.2003-0567
[8]Rossi, L., et al. (2017) Clinopathologic characteristics of endometrial cancer in Lynch syndrome: a French multicenter study. Int J Gynecol Cancer. 27(5): 953-960. doi: 10.1097/IGC.0000000000000985
[9] Sasso et al. (2019) Estradiol and progesterone regulate proliferation and apoptosis in colon cancer. Endocrine Connections. 8(3): 217-229. doi: 10.1530/EC-18-0374
Images:
Header: static01.nyt.com/images/2018/03/25/opinion/sunday/25reich/25reich-articleLarge.jpg?quality=75&auto=webp&disable=upscale
Mouse: www.jax.org/-/media/jaxweb/images/jaxmiceandservices/mice/datasheets/005557.jpgh=224&w=398&la=en&hash=5B303DE5BCB4FE9A45E68BF3C9814301FB172CDD
Endometrius: us.123rf.com/450wm/tpimovit/tpimovit1809/tpimovit180900120/110860581-stock-vector-vector-isolated-illustration-of-female-reproductive-system-anatomy-uterus-cervix-ovary-fallopian-tub.jpg?ver=6
Estrogen:upload.wikimedia.org/wikipedia/commons/thumb/0/00/Estradiol.svg/1200px-Estradiol.svg.png
| williams_finaldraft.pdf |
